什么是色谱分析法_什么是色谱分析的原理
色谱法是实验室非常重要又常用的分析方法,由于色谱分析可以连续对样品进行浓缩、分离、提纯及测定,使其成为每一个分析工作者普遍采用的分析、检测手段,并已广泛应用于各个行业中,可以说只要有分析任务的地方都在使用色谱分析法。今天咱们补充基础知识哦。
色谱法的创始人是俄国的植物学家茨维特( M.Tswett)。1906年,俄国植物学家茨维特发表了他的实验结果:为了分离植物色素,他将含有植物色素的石油醚提取液倒入装有碳酸钙粉末的玻璃管中,并用石油醚自上而下淋洗,由于不同的色素在碳酸钙颗粒表面的吸附力不同,随着淋洗的进行,不同色素向下移动的速度不同,从而形成一圈圈不同颜色的色带,使各色素成分得到了分离。他将这种分离方法命名为色谱法(chromatography)。

伟大的茨维特
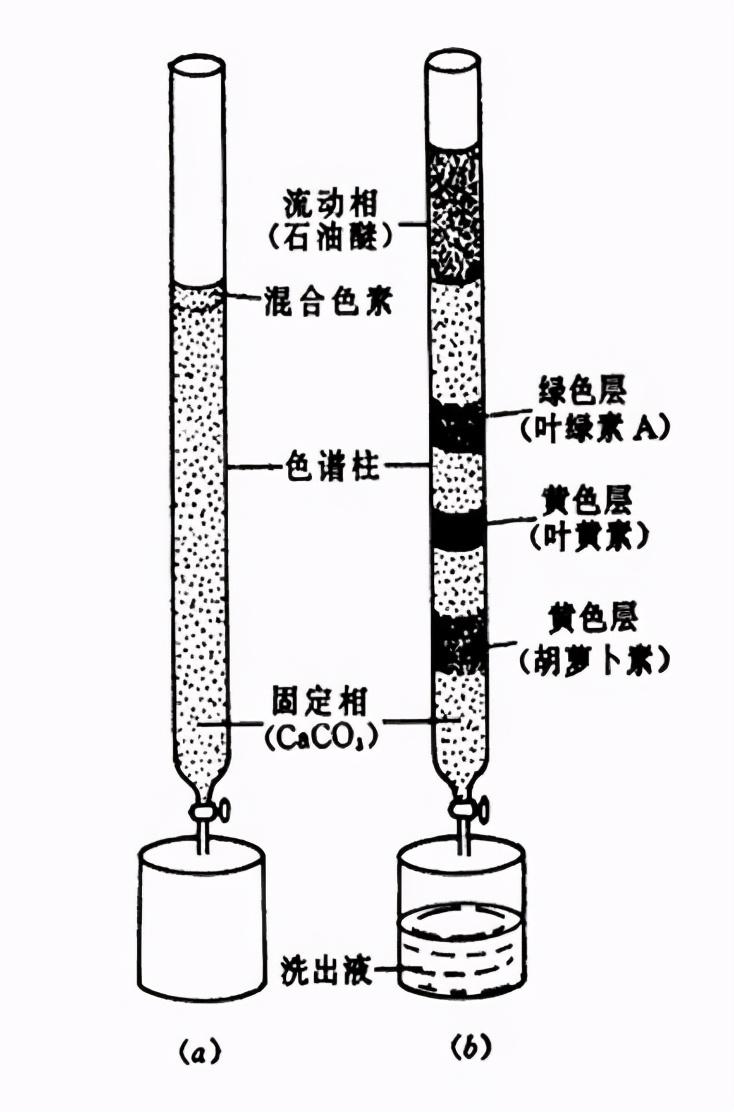
植物叶色素的分离实验
☞茨维特实验图示
√ 色谱柱—玻璃柱
√ 固定相—CaCO3颗粒
√ 流动相—石油醚
随着分离手段的不断发展,越来越多的无色物质成为被分离的对象,色谱也渐渐失去了“色”的含义,但这个名称却沿用至今。
色谱分析法(Chromatography)简称色谱法或层析法,是一种物理或物理化学分离分析方法,该法利用某一特定的色谱系统(薄层色谱、高效液相色谱或气相色谱等系统)进行混合物中各组分的分离分析,主要用于分析多组分样品。
▪ 固定相:在色谱分离中固定不动,对样品产生保留的一相。
▪ 流动相:带动样品向前移动的另一相。
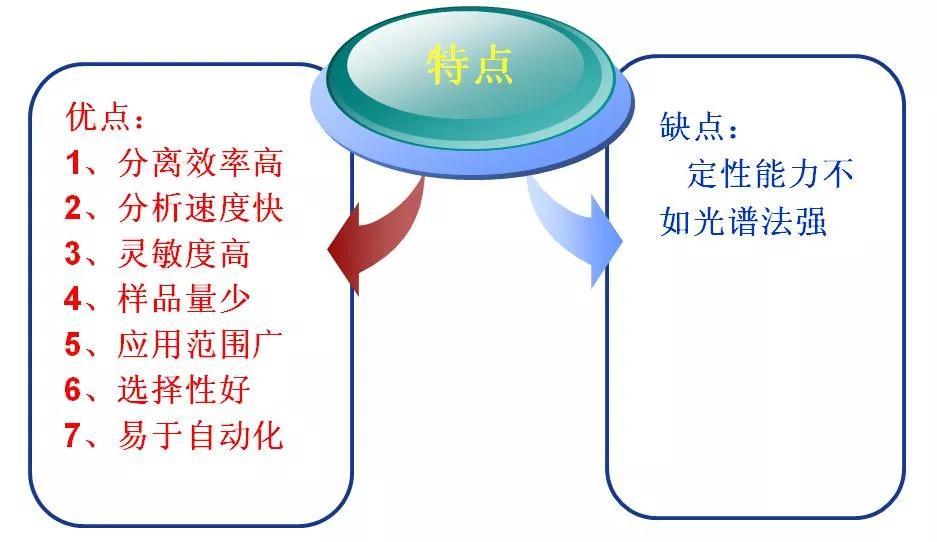
分类
1、按两相物理状态分类
▪ 流动相:气相色谱、液相色谱、超临界流体色谱
▪ 固定相:气-固、气-液;液-固、液-液
2、按固定相的形式分类
▪ 柱色谱:填充柱色谱、毛细管柱色谱、微填充柱色谱、制备色谱
▪ 平面色谱:纸色谱、薄层色谱、高分子膜色谱
3、按分离机制分类
▪ 吸附色谱:根据不同组分在吸附剂上的吸附和解吸能力的大小而分离
▪ 分配色谱:根据不同组分在固定液中溶解度的大小而分离
▪ 分子排阻色谱:依据分子体积大小不同进行分离ln离子交换色谱:不同组分对离子交换树脂的亲和力不同而分离
▪ 亲和色谱:利用生物大分子之间的存在的专一的特殊亲和力进行分离
▪ 毛细管电泳:依据各组分淌度和(或)分配行为的差异进行分离
▪ 手性色谱:用于手性药物的分离分析,可分为三类:手性衍生化试剂法;手性流动相添加剂法;手性固定相拆分法
基本术语
1、色谱法的基本术语
检测色谱分离后组分的响应信号对时间作图得到的曲线称为色谱图。

色谱流出曲线


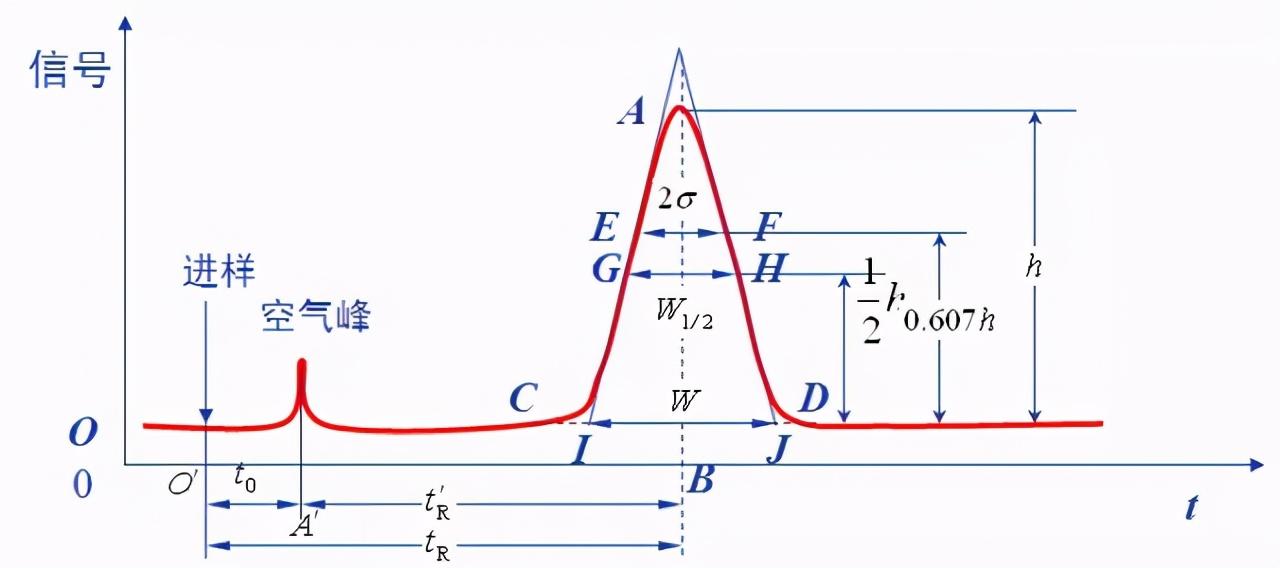

▪基线:在一定色谱条件下,仅有流动相通过检测器系统时所产生的信号的曲线,称为基线,如图中ot线。实验条件稳定时,基线是一条平行于横轴的线;基线反映仪器(主要是检测器)的噪声随时间的变化。
▪峰高:色谱峰顶点与基线之间的垂直距离,以h表示,如图AB’线。
▪区域宽度:色谱峰的区域宽度直接与分离效率有关。描述色谱峰宽的方法有三种:标准偏差σ、峰宽W、半峰宽W1/2。
▪标准偏差(σ):σ为正态分布曲线上两拐点间距离之半,σ值的大小表示组分离开色谱柱的分散程度。σ值越大,流出的组分越分散,分离效果变差;反之,流出组分集中,分离效果好。
▪峰宽W:通过色谱峰两侧的拐点作切线,在基线上的截距称为峰宽,或称基线宽度,也可用W表示,如图IJ的距离。根据正态分布的原理 ,可证得峰宽和标准差的关系是W=4σ。
▪半峰宽W1/2:峰高一半处的峰宽称为半峰宽,如图GH的距离。W1/2=2.355σ,W=1.699W1/2。
W1/2、W都是由σ派生而来的,除用它们衡量柱效外,还用于计算峰面积。半峰宽测量较方便,最为常用。
2、色谱保留值、相对保留值与保留指数
▪保留值
是用来描述样品组分在色谱柱中保留程度的参数,并作为色谱定性的指标。其表示方法有:

▪相对保留值
又称分离因子、分配系数比或相对容量因子,即在一定色谱条件下,被测组分的调整保留时间(体积)与标准物的调整保留时间(体积)之比。

采用相对保留值是为了消除某些操作条件,如流速和固定液流失等,对保留值的影响。相对保留值中标准物可以是被测样品中的某一组分,也可以是人为加入的某一化合物。
▪保留指数
I为待测物质i在某固定液X上的保留指数,选取两个正构烷烃作为基准物质,其中一个的碳数为N,另一个为N+n,它们的调整保留时间分别为t’R(N)和 t’R(N+n),使待测物质i的调整保留时间t’R(i)恰好于两者之间,即t’R(N)<t’R(i)<t’R(N+n)。将含物质i和所选的两个正构烷烃的混合物注入其固定液X的色谱柱,在一定温度条件下绘制色谱图。
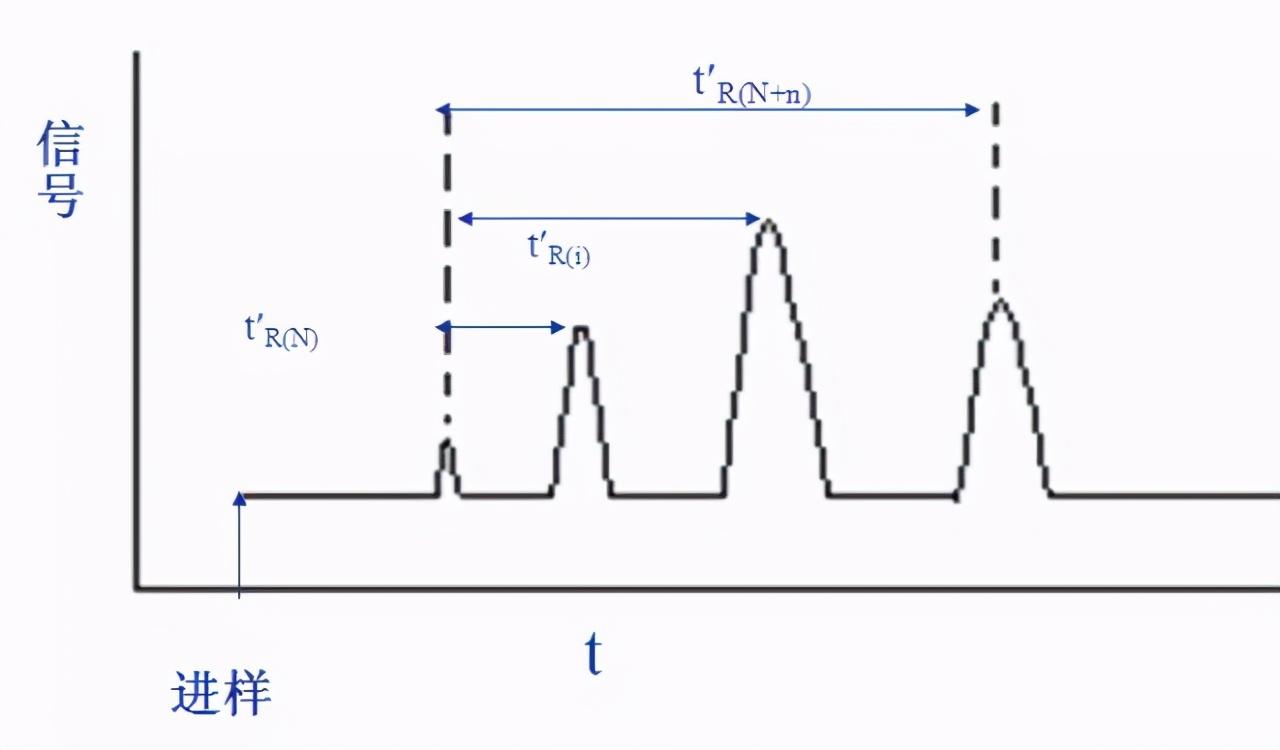
保留指数的计算式为:
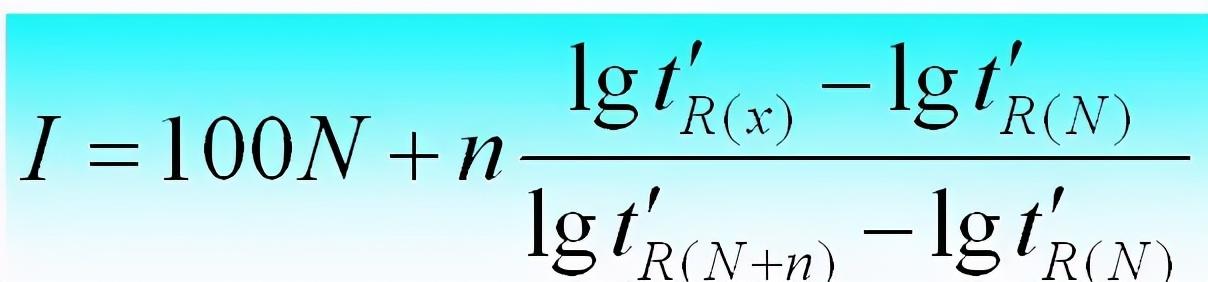
当相邻两个正构烷的碳数差值n=1时,保留指数或简化为:
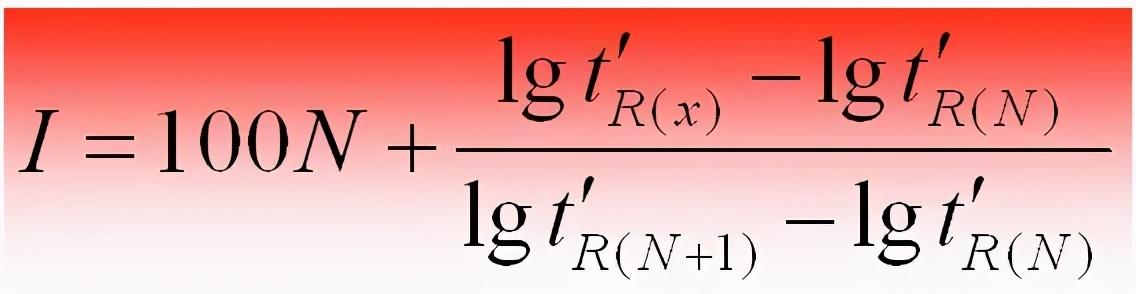
3、容量因子与分配系数
(1)容量因子(k)
在平衡状态下,组分在固定相(s)与流动相(m)中的质量之比,称为容量因子。公式如下:

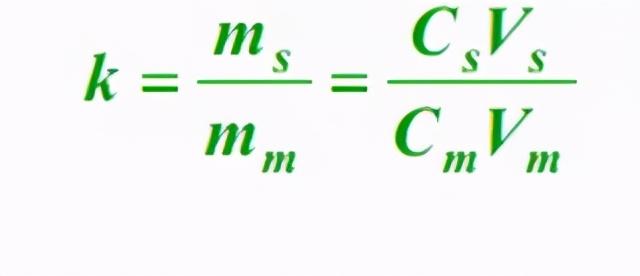
(2)分配系数(K)
在平衡状态下,组分在固定相(s)与流动相(m)中的浓度之比,称为分配系数。公式如下:

☞ K与k的关系:

☞ 保留值与容量因子及分配系数的关系
色谱分离是基于固定相对试样中各组分的吸附或溶解能力强弱的不同,而这种吸附或溶解能力的强弱可定量地用分配系数K(或容量因子k)值的大小来表示。吸附或溶解能力强的组分分配系数(或容量因子)大,保留时间长;反之,吸附或溶解能力弱的组分分配系数小,保留时间短。
基本理论
主要包括塔板理论和速率理论。
1、塔板理论
(1)提出——热力学理论
▪ 始于马丁(Martin)和辛格(Synge)提出的塔板模型。
▪ 分馏塔:在塔板上多次气液平衡,按沸点不同而分离。
▪ 色谱柱:组分在两相间的多次分配平衡,按分配系数不同而分离。
(2)假设
(1)色谱柱内存在许多塔板,组分在塔板间隔(即塔板高度)内可以很快达到分配平衡。
(2)流动相进入色谱柱,不是连续的而是脉动式的,即每次通过为一个塔板体积。
(3)样品加在每个塔板上,样品沿色谱柱轴方向的扩散可以忽略。
(4)在所有塔板上分配系数相等,与组分的量无关。即分配系数在各塔坂上是常数。
(3)原理

塔板理论示意图
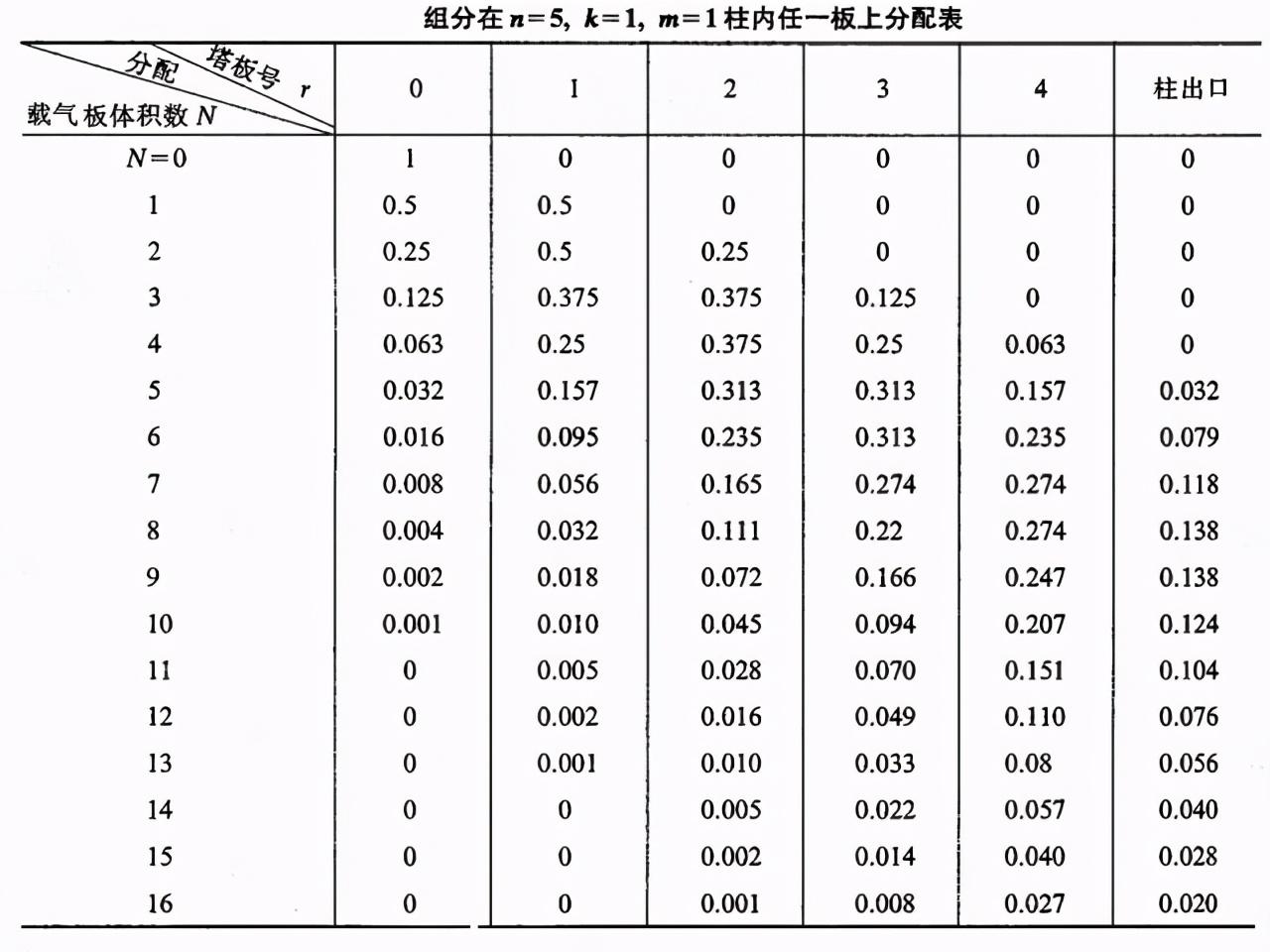
▪ 开始时,若有单位质量,即m=1(例1mg或1μg)的某组分加到第0号塔板上,分配平衡后,由于k=1,即ns=nm故nm=ns=0.5。
▪ 当一个板体积(lΔV)的载气以脉动形式进入0号板时,就将气相中含有nm部分组分的载气顶到1号板上,此时0号板液相中ns部分组分及1号板气相中的nm部分组分,将各自在两相间重新分配。故0号板上所含组分总量为0.5,其中气液两相各为0.25而1号板上所含总量同样为0.5.气液相亦各为0.25。
▪ 以后每当一个新的板体积载气以脉动式进入色谱柱时,上述过程就重复一次(见下表)。
(4)色谱流出曲线方程

(5)色谱柱效参数

理解:在tR一定时,W或W1/2越小(即峰越窄),理论板数n越大, 理论板高越小,柱的分离效率越高。有效理论塔板neff也同此理。因此,理论塔板数是评价柱效能的指标。
(6)特点及不足

2、速率理论
1956年荷兰学者VanDeemter等人吸收了塔板理论的概念,并把影响塔板高度的动力学因素结合起来,提出了色谱过程的动力学理论——速率理论,导出了Van Deemter方程。它把色谱过程看作一个动态非平衡过程,研究过程中的动力学因素对峰展宽(即柱效)的影响。
后来Giddings和Snyder等人在VanDeemter方程(后称气相色谱速率方程)的基础上,根据液体与气体的性质差异,提出了液相色谱速率方程(即Giddings方程)。
(1)Van Deemter方程


(2) Giddings方程

定量与定性分析
(一)定性分析
色谱定性分析就是要确定各色谱峰所代表的化合物。由于各种物质在一定的色谱条件下均有确定的保留值,因此保留值可作为一种定性指标。目前各种色谱定性方法都是基于保留值的。
但是不同物质在同一色谱条件下,可能具有相似或相同的保留值,即保留值并非专属的。因此仅根据保留值对一个完全未知的样品定性是困难的。如果在了解样品的来源、性质、分析目的的基础上,对样品组成作初步的判断,再结合下列的方法则可确定色谱峰所代表的化合物。
1、利用纯物质对照定性
在一定的色谱条件下,一个未知物只有一个确定的保留时间。因此将已知纯物质在相同的色谱条件下的保留时间与未知物的保留时间进行比较,就可以定性鉴定未知物。若二者相同,则未知物可能是已知的纯物质;不同,则未知物就不是该纯物质。
纯物质对照法定性只适用于组分性质已有所了解,组成比较简单,且有纯物质的未知物。
2、相对保留值法

在某一固定相及柱温下,分别测出组分i和基准物质s的调整保留值,再按上式计算即可。用已求出的相对保留值与文献相应值比较即可定性。
3、加入已知物增加峰高法
当未知样品中组分较多,所得色谱峰过密,用上述方法不易辨认时,或仅作未知样品指定项目分析时均可用此法。
首先作出未知样品的色谱图,然后在未知样品加入某已知物,又得到一个色谱图。峰高增加的组分即可能为这种已知物。
4、保留指数定性法
保留指数表示物质在固定液上的保留行为,是目前GC中使用最广泛并被国际上公认的定性指标。它具有重现性好、标准统一及温度系数小等优点。
保留指数仅与固定相的性质、柱温有关,与其它实验条件无关。其准确度和重现性都很好。只要柱温与固定相相同,就可应用文献值进行鉴定,而不必用纯物质相对照。
(二)定量分析


1、峰面积测量方法
峰面积是色谱图提供的基本定量数据,峰面积测量的准确与否直接影响定量结果。对于不同峰形的色谱峰采用不同的测量方法。
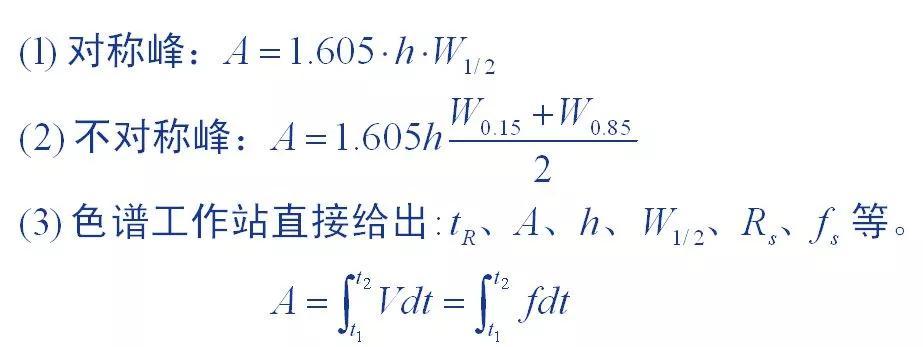
2、定量校正因子




3、定量计算方法
(1)面积归一化法

(2)外标法
以待测组分纯品为对照物,与试样中待测组分的响应信号相比较进行定量的方法。
(3)内标法
所谓内标法,是将一定量的纯物质作为内标物加入到待测物的标准溶液和样品溶液中,再进行分析测定的方法。
(4)标准加入法